
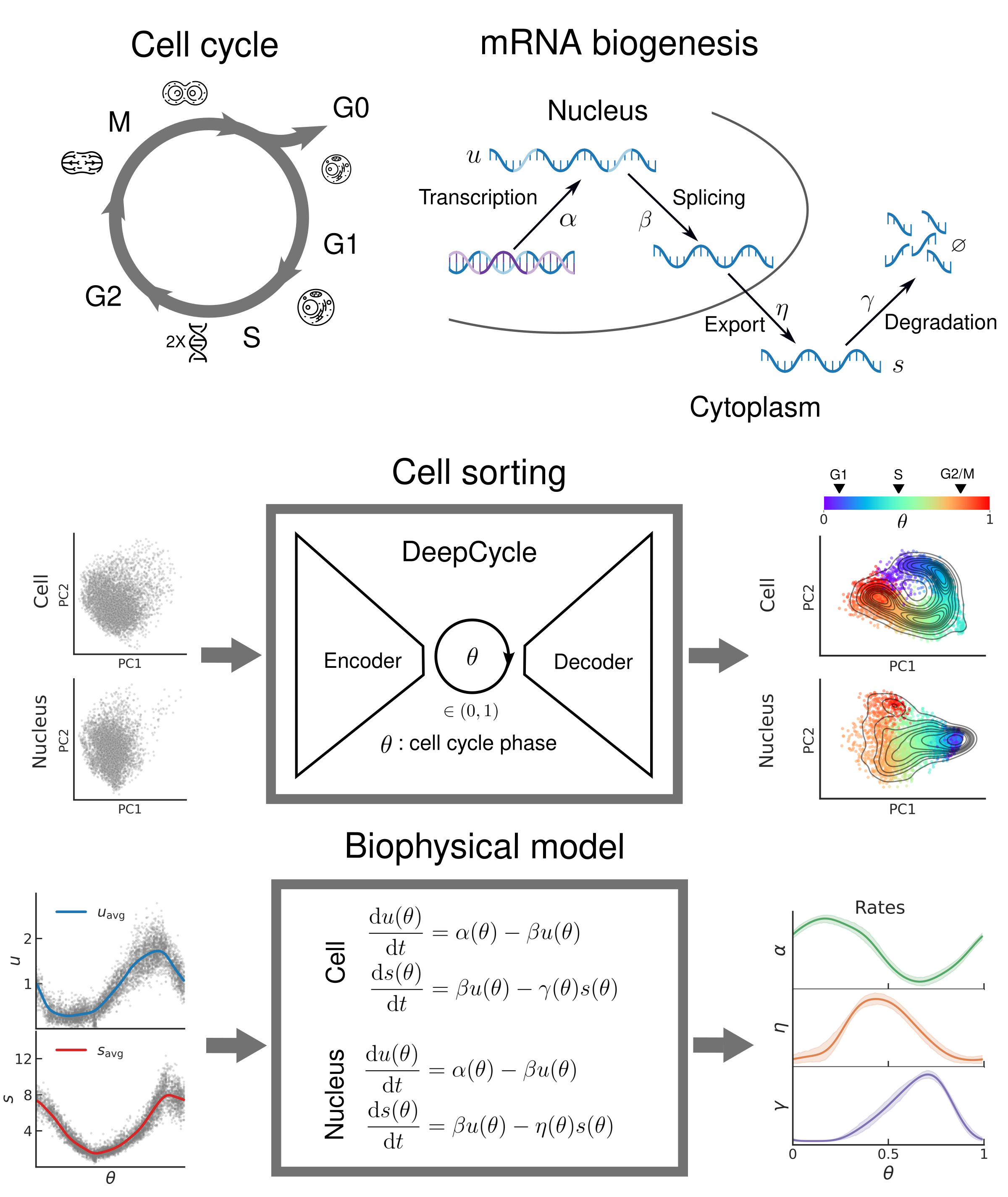
Oscillatory dynamics of mRNA biogenesis and chromatin accessibility during the cell cycle
The cell cycle is a highly regulated process that ensures the accurate replication and transmission of genetic information from one generation of cells to the next. It is a fundamental biological process and plays a crucial role in development, growth, and maintenance of all living organisms, and its dysregulation can lead to a number of pathologies such as autoimmune diseases, neurodegenerative diseases, and cancer. In this work we present a novel approach to study the gene expression and chromatin accessibility dynamics during the cell cycle in mouse embryonic stem cells. To achieve this, we combined high-depth single-cell multiomic sequencing, biophysical modeling, and advanced deep learning techniques. First, we used DeepCycle, a deep learning tool that assigns a cell cycle phase to every cell based on the spliced and unspliced mRNA reads from scRNA-seq. We then developed a biophysical model that describes the dynamics of gene-specific mRNA synthesis, export, and degradation rates during the cell cycle. The key feature of single-cell multiomic sequencing is that it simultaneously gives readouts for gene expression as well as chromatin accessibility in the same cells. By applying our approach to these data we showcase first-of-its-kind exploration of chromatin accessibility during the cell cycle at high temporal resolution. We believe that our work would be pivotal in formulating a coherent quantitative description of mRNA biogenesis.
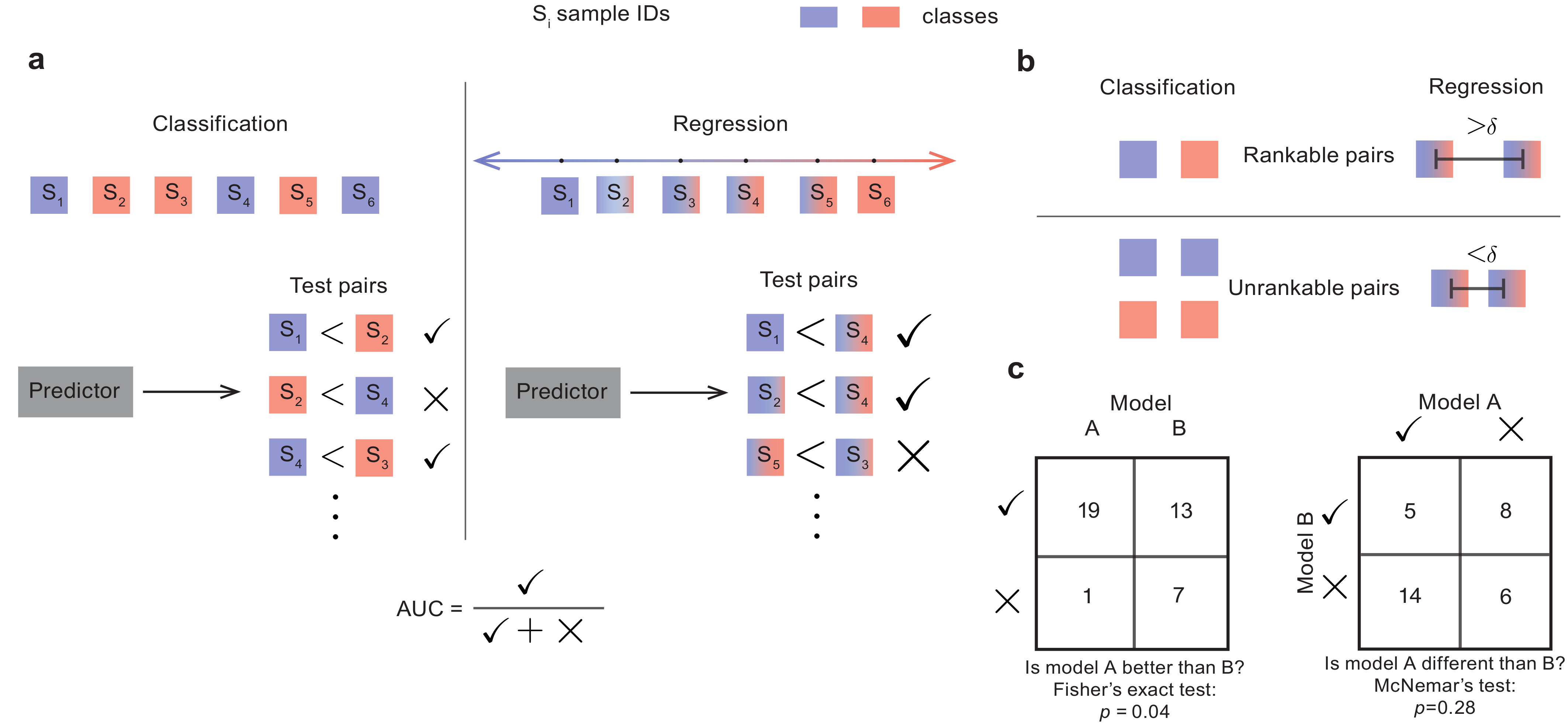
Paired evaluation for improvevd estimates of performance of machine learning models
The true accuracy of a machine learning model is a population-level statistic that cannot be observed directly. In practice, predictor performance is estimated against one or more test datasets, and the accuracy of this estimate strongly depends on how well the test sets represent all possible unseen datasets. We present a simple approach, paired evaluation, for increasing the robustness of performance evaluation by systematic pairing of test samples, and use it to evaluate predictors of drug response in breast cancer cell lines and of disease severity in patients with Alzheimer’s Disease. Our results demonstrate that the choice of test data can cause variations in estimates of performance and that paired evaluation makes it possible to identify outliers, improve the accuracy of performance estimates in the presence of known confounders, and assign statistical significance when comparing machine learning models.
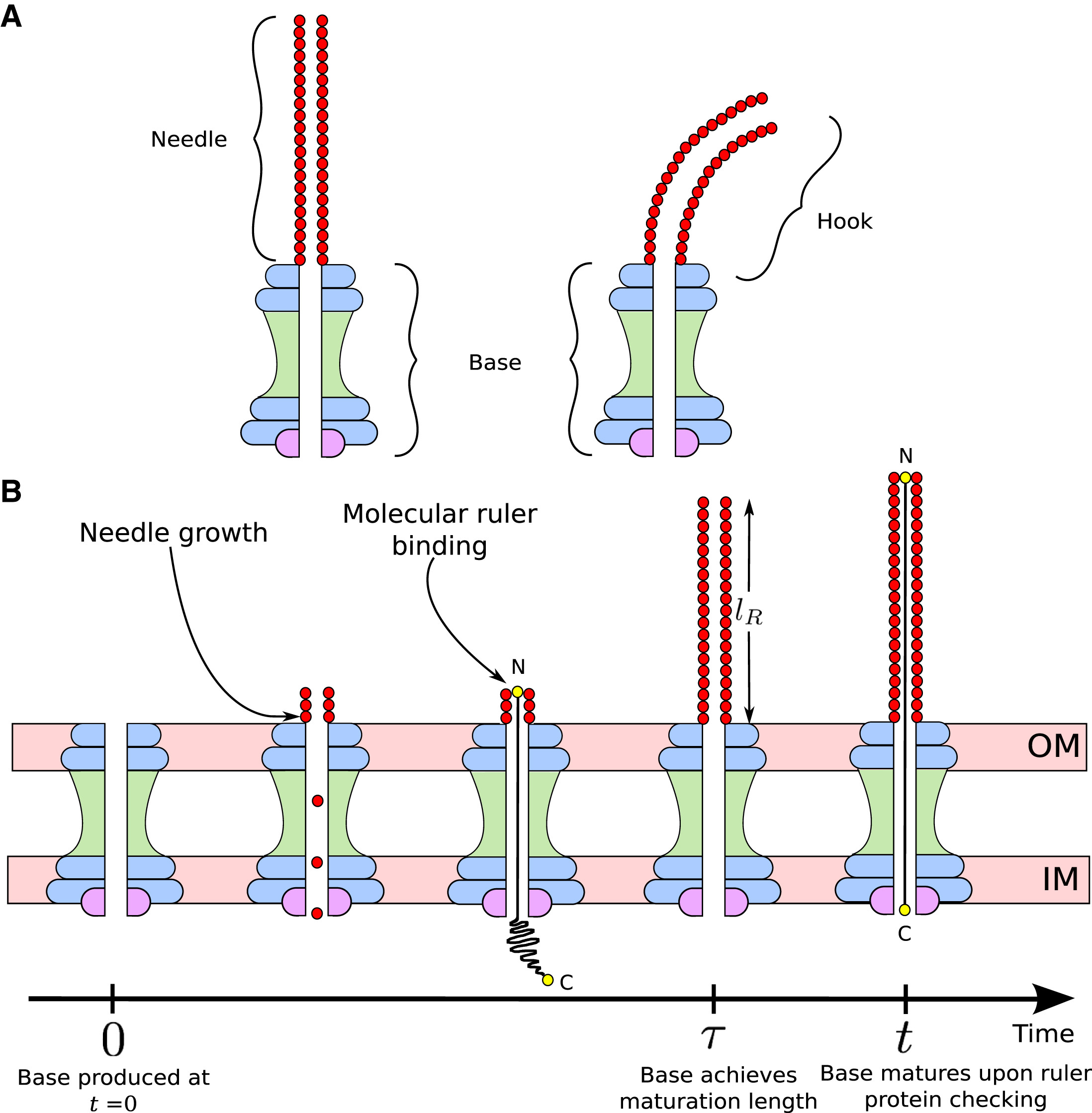
Mathematical models for length control in bacterial structures
Bacterial cells construct many structures, such as the flagellar hook and the type III secretion system (T3SS) injectisome, that aid in crucial physiological processes.The length of these channels must be highly regulated in order for these structures to perform their intended functions. There are two leading mechanisms for length regulation in the flagellar hook and T3SS needle: the substrate switching mechanism, in which the length is controlled by assembly of an inner rod, and the ruler mecchanism, in which a molecular ruler controls the length. Using principles from dynamical systems, probability theory, and Gillespie algorithm, we developed mathematical models for these mechanisms. Our models suggest that for the susbtrate switching mechanism the variance in length distribution of these structures would scale quadractically with the average length, whereas for the ruler mechanism the variance would scale linearly with the average length. These findings shed new light on the trade-offs that may have led to the evolution of different length control mechanisms in different bacterial species.
| Ruler mechanism | Substrate switching mechanism |
|---|---|
 | 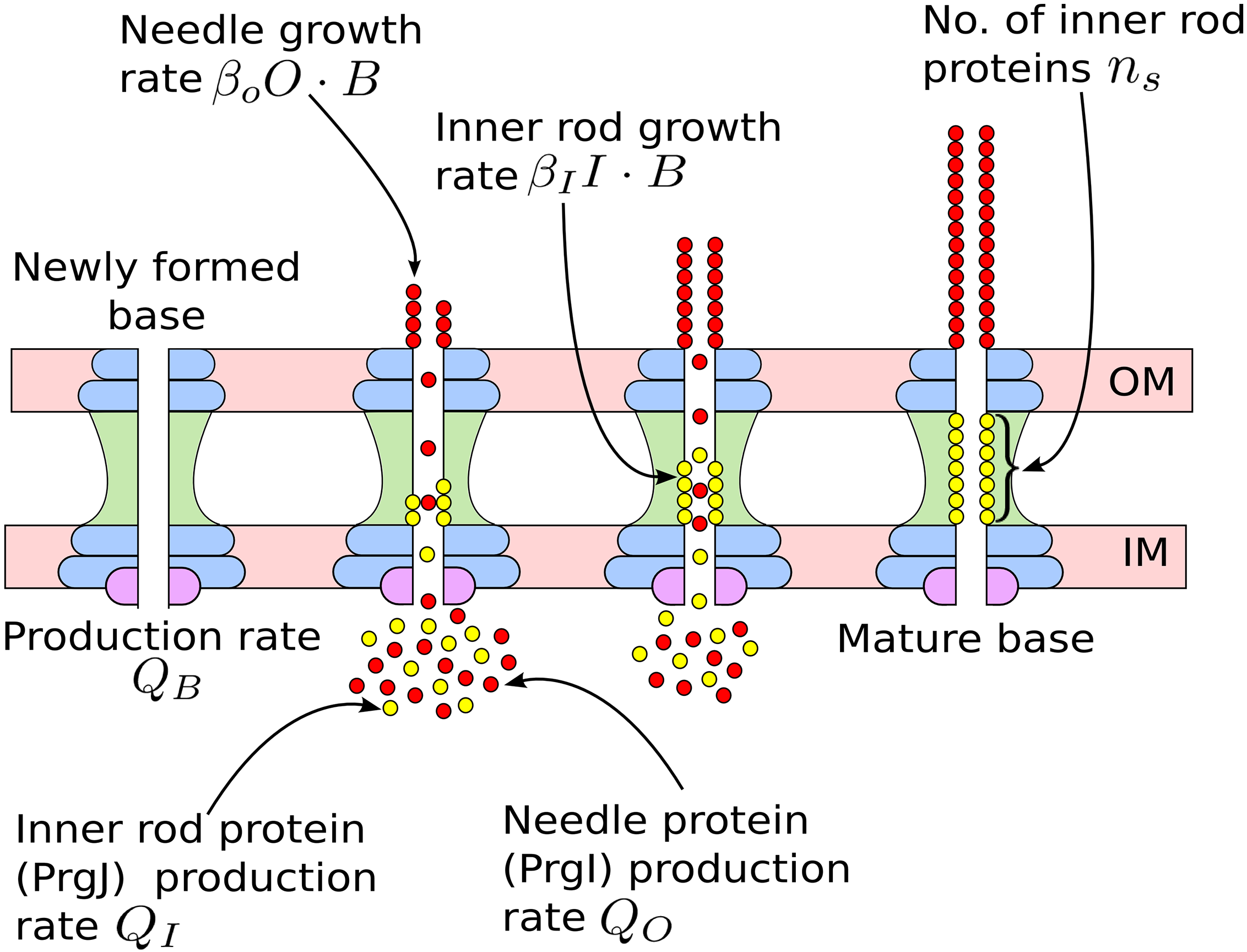 |